Cell Embeddings with BrainBeacon#
This tutorial shows how to obtain cell embeddings from a pretrained BrainBeacon checkpoint (Heffel2024 MERFISH as an example).
Users can run BrainBeacon on their own AnnData by replacing:
adata_pathspecies/assay(if needed)gene_mean_path(assay/platform-specific prior stats)
import os
import torch
from brainbeacon.pipeline.cell_embedding import run_bbcellformer_pipeline
from brainbeacon.configs.config import resolve_path
from brainbeacon.configs.config_train import config_train
Device setup#
# NOTE: If you want to pin a specific GPU, uncomment and edit the line below.
# os.environ["CUDA_VISIBLE_DEVICES"] = "0"
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
print(f"Using device: {device}")
if device.type == "cuda":
print(f"Using GPU: {torch.cuda.get_device_name(torch.cuda.current_device())}")
Using device: cuda
Using GPU: Tesla V100-SXM2-16GB
Paths and dataset config#
Example dataset (Heffel2024 MERFISH, GEO: GSE213950)
GEO page: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE213950
File:
GSE213950_HPC_spatial_MERFISH_01132024.h5ad.gz
If you don’t have the .h5ad locally yet, you can download the file from GEO
and place it under the path you want (e.g. BrainBeacon/data/.../processed/),
then update adata_path accordingly.
BASE_DIR = "/raid/zhangchengming/BrainBeacon-master"
dataset_name = "heffel2024"
species = "human"
assay = "merfish"
input_data_dir = os.path.join(
BASE_DIR, "data", "MERFISH_Human_Heffel2024Temporally3D", "processed"
)
adata_path = os.path.join(input_data_dir, "Heffel2024Temporally3D.h5ad")
gene_dict_path = resolve_path("GENE_DICT_PATH")
prior_dir = resolve_path("PRIOR_DIR")
gene_mean_path = os.path.join(prior_dir, "merfish_gene_nonzero_means.npy")
print("adata_path:", adata_path)
print("gene_dict_path:", gene_dict_path)
print("gene_mean_path:", gene_mean_path)
assert os.path.exists(adata_path), f"adata_path not found: {adata_path}"
assert os.path.exists(gene_dict_path), f"gene_dict_path not found: {gene_dict_path}"
assert os.path.exists(gene_mean_path), f"gene_mean_path not found: {gene_mean_path}"
adata_path: /raid/zhangchengming/BrainBeacon-master/data/MERFISH_Human_Heffel2024Temporally3D/processed/Heffel2024Temporally3D.h5ad
gene_dict_path: /raid/zhangchengming/BrainBeacon-master/prior_knowledge/model_h5ad_1211.h5ad
gene_mean_path: /raid/zhangchengming/BrainBeacon-master/prior_knowledge/merfish_gene_nonzero_means.npy
Pretrained checkpoints#
pretrain_dir = resolve_path("PRETRAIN_DIR")
bb_ckpt_name = "epoch_0_step_800000_0.33B.pt"
bb_ckpt_path = os.path.join(pretrain_dir, bb_ckpt_name)
cellplm_ckpt_path = os.path.join(
BASE_DIR,
"downstream_tasks", "train_cellformer", "epoch_0_step_800000_0.33B",
"cellformer_epoch100.pt"
)
print("bb_ckpt_path:", bb_ckpt_path)
print("cellplm_ckpt_path:", cellplm_ckpt_path)
assert os.path.exists(bb_ckpt_path), f"bb_ckpt_path not found: {bb_ckpt_path}"
assert os.path.exists(cellplm_ckpt_path), f"cellplm_ckpt_path not found: {cellplm_ckpt_path}"
bb_ckpt_path: /raid/zhangchengming/BrainBeacon-master/pretrained/epoch_0_step_800000_0.33B.pt
cellplm_ckpt_path: /raid/zhangchengming/BrainBeacon-master/downstream_tasks/train_cellformer/epoch_0_step_800000_0.33B/cellformer_epoch100.pt
Output naming#
method_name = "brainbeacon"
output_prefix = f"{dataset_name}_{method_name}"
output_dir = os.path.join(
BASE_DIR, "downstream_tasks", "cell_clustering", "outputs", dataset_name, method_name
)
os.makedirs(output_dir, exist_ok=True)
print("output_dir:", output_dir)
print("output_prefix:", output_prefix)
output_dir: /raid/zhangchengming/BrainBeacon-master/downstream_tasks/cell_clustering/outputs/heffel2024/brainbeacon
output_prefix: heffel2024_brainbeacon
Run pipeline#
n_hvg = 1000
cd_weight = 0.02
do_fit = False
fit_epochs = 10
adata = run_bbcellformer_pipeline(
adata_path=adata_path,
specie=species, # pipeline uses "specie" as argument name
assay=assay,
gene_dict_path=gene_dict_path,
gene_mean_path=gene_mean_path,
bb_ckpt_path=bb_ckpt_path,
cellplm_ckpt_path=cellplm_ckpt_path,
output_dir=output_dir,
output_prefix=output_prefix,
config_train=config_train,
n_hvg=n_hvg,
cd_weight=cd_weight,
use_hvg=True,
use_batch=True,
use_spatial=True,
weight_mode="expression",
force_tokenize=False,
do_fit=do_fit,
fit_epochs=fit_epochs,
device=device,
)
print("Pipeline finished.")
print("obsm keys:", list(adata.obsm.keys()))
print("obs columns (first 30):", list(adata.obs.columns)[:30])
Tokenized data found (6 .parquet, 6 dirs). Skipping tokenization.
Skipping BB inference. Found existing file: /raid/zhangchengming/BrainBeacon-master/downstream_tasks/cell_clustering/outputs/heffel2024/brainbeacon/heffel2024_brainbeacon_bb_embeddings.npz
[INFO] Using explicitly provided CellFormer checkpoint: /raid/zhangchengming/BrainBeacon-master/downstream_tasks/train_cellformer/epoch_0_step_800000_0.33B/cellformer_epoch100.pt
********** gene list size: 92076 **********
********** loading skip parameters: set() **********
After filtering, 298 genes remain.
Model saved to /raid/zhangchengming/BrainBeacon-master/downstream_tasks/cell_clustering/outputs/heffel2024/brainbeacon/heffel2024_brainbeacon_cellformer.pt
Embeddings saved to /raid/zhangchengming/BrainBeacon-master/downstream_tasks/cell_clustering/outputs/heffel2024/brainbeacon/heffel2024_brainbeacon_embeddings.npz
Pipeline finished.
obsm keys: ['H3K9me3', 'K27Ac', 'LamA', 'Nup98', 'Pol2PSer2', 'Sc35', 'X_diffmap', 'X_fov', 'X_h_score', 'X_norm', 'X_pca', 'X_raw', 'X_spatial', 'X_umap', 'blank', 'spatial', 'bb_emb', 'X_emb', 'X_pred']
obs columns (first 30): ['volm', 'x_um_abs', 'y_um_abs', 'zc', 'xc', 'yc', 'leiden', 'region', 'L1', 'dpt_pseudotime', 'final_anno_v3', 'hpc_regional', 'fimbria_regional', 'hpcRG', 'fimbriaRG', 'ventricular_regional', 'ventricularRG', 'refined_volume', 'cell_label', 'slice', 'species', 'split', 'platform', 'valid_split', 'batch', 'x_FOV_px', 'y_FOV_px']
UMAP (colored by Leiden)#
import scanpy as sc
# Use X_emb for downstream UMAP (as requested)
assert "X_emb" in adata.obsm, "Expected `adata.obsm['X_emb']` for UMAP, but it was not found."
# Compute UMAP if not already present
if "X_umap" not in adata.obsm:
sc.pp.neighbors(adata, use_rep="X_emb")
sc.tl.umap(adata)
# Compute Leiden if not already present
if "leiden" not in adata.obs:
sc.tl.leiden(adata, resolution=1.0)
sc.pl.umap(
adata,
color="leiden",
legend_loc="right margin",
title="UMAP (Leiden)",
show=True,
)

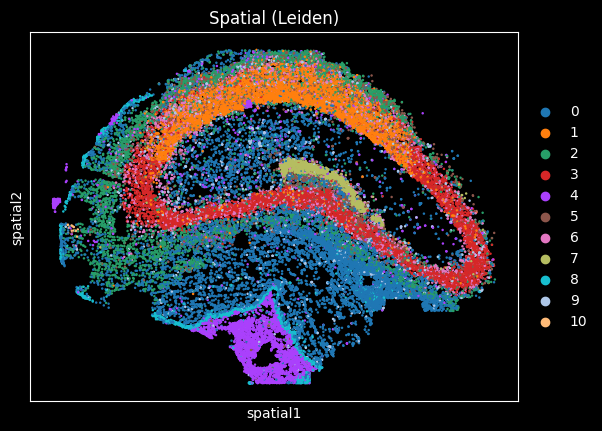
Spatial plot (optional)#
If your AnnData contains spatial coordinates in adata.obsm['spatial'] (Scanpy convention),
you can visualize clusters in tissue space:
# Spatial plot (optional)
import numpy as np
import scanpy as sc
if "spatial" in adata.obsm:
spatial = adata.obsm["spatial"]
# Convert DataFrame -> numpy, or keep numpy as-is
if hasattr(spatial, "to_numpy"):
spatial = spatial.to_numpy()
# Ensure (n_cells, 2)
spatial = np.asarray(spatial)
if spatial.ndim != 2 or spatial.shape[1] < 2:
raise ValueError(f"adata.obsm['spatial'] must be (n_cells, >=2), got {spatial.shape}")
# Keep only first 2 dims
adata.obsm["spatial"] = spatial[:, :2]
sc.pl.spatial(
adata,
color="leiden",
legend_loc="right margin",
spot_size=40,
title="Spatial (Leiden)",
show=True,
)
else:
print("Skip spatial plot: `adata.obsm['spatial']` not found.")
Notes for users#
Cell embeddings are stored in
adata.obsm['X_emb'].This notebook uses
X_embto compute UMAP and colors byleiden.To run on another dataset, replace:
adata_pathspecies/assay(if needed)gene_mean_path(platform-specific prior stats)